Published in Volume 114, Issue 11 (December 1, 2004)
J Clin Invest. 2004;114(11):1538–1540. doi:10.1172/JCI23705.
Copyright © 2004, American Society for Clinical Investigation
J Clin Invest. 2004;114(11):1538–1540. doi:10.1172/JCI23705.
Copyright © 2004, American Society for Clinical Investigation
3 citations have been reported for this article.
Commentary
PPARs in atherosclerosis: the clot thickens
Howard Hughes Medical Institute and Department of Pathology and Laboratory Medicine, UCLA, Los Angeles, California, USA.
Address correspondence to: Peter Tontonoz, Howard Hughes Medical Institute, UCLA, Box 951662, Los Angeles 90095-1662, California, USA. Phone: (310) 206-4546; Fax: (310) 267-0382; E-mail: ptontonoz@mednet.ucla.edu.Published December 1, 2004
Cardiovascular disease is the leading cause of morbidity and mortality in Western countries. Previous studies have highlighted the beneficial effects of PPARγ activators on cardiovascular disease; however, the role of other PPAR family members in atherosclerosis is less clear. A report in this issue of the JCI expands our understanding of PPARs in vascular biology and highlights the potential use of multiple PPAR agonists to limit lipid accumulation in macrophages.
PPARs and atherosclerosis: nuclear receptor action in the artery wall
Atherosclerosis is a chronic inflammatory disease as well as a disorder of lipid metabolism (1). The accumulation of cholesterol-rich lipoproteins in the artery wall results in the recruitment of circulating monocytes, their adhesion to the endothelium, and their differentiation into tissue macrophages. Lipid-loaded macrophages play an important role in the production of chemokines, cytokines, and reactive oxygen species in the early stages of lesion formation. Therefore mechanisms that limit macrophage cholesterol accumulation and/or prevent the production of inflammatory mediators all have the potential to inhibit lesion development.
The PPAR family is comprised of 3 different proteins: PPARα, PPARβ (also referred to as δ), and PPARγ (2). Natural ligands for these receptors include fatty acids and oxidized fatty acids. The relevance of PPAR pathways to metabolic disease is underscored by the use of the fibrates (PPARα agonists) and thiazolidinediones (PPARγ agonists) to treat hyperlipidemia and type 2 diabetes, respectively. The expression of PPARs in cells of the artery wall has prompted a number of investigations into the effects of PPAR agonists on atherosclerosis in mice (3). Studies on PPARγ are in general agreement that activation of this receptor in the artery wall is beneficial (4–6). However, studies using PPARα- and PPARβ-knockout mice have yielded more complex results. Transplantation of bone marrow lacking PPARβ has been reported to reduce atherosclerosis in apoE–/– mice (7). Similarly, mice lacking both PPARα and apoE were shown to develop fewer lesions (8). On the other hand, intervention studies using PPARα agonists have suggested antiatherogenic effects in mice (9), and the Veterans Affairs High-Density Lipoprotein Intervention Trial showed a clear reduction in cardiovascular events in patients taking gemfibrozil (10). The impact of PPARβ agonists on atherosclerosis is unknown, although GW1516 was shown to have beneficial effects on plasma lipid profiles in obese rhesus monkeys (11).
PPARs are known to influence pathways for both lipid uptake and efflux in macrophages. PPARγ promotes CD36 expression (12), and both PPARα and PPARγ induce expression of liver X receptor α (LXRα) and ABCA1 (4, 13) (Figure 1). However, the ability of PPARs to control LXRα expression is much more prominent in human cells than in murine cells, raising the possibility that additional pathways are involved in the beneficial effects of PPARs observed in murine models. In addition to their effects on lipid metabolism, PPAR activators also inhibit inflammatory gene expression in cultured macrophages (14). Glass and colleagues have further shown that treatment of LDL receptor–deficient (LDLR–/–) mice with PPARγ agonists reduced the expression of inflammatory mediators (5). Thus inhibition of inflammation represents a second mechanism by which PPAR activation might influence atherogenesis.
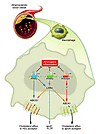 Figure 1
Figure 1PPAR signaling pathways influence macrophage gene expression and foam-cell formation. Ligand activation of PPARα and PPARγ, but not PPARβ/δ, inhibits the development of atherosclerosis in LDLR_/_ mice. Both systemic and local mechanisms might contribute to these beneficial effects. Previous studies have suggested that PPARα and PPARγ increase LXRα expression in macrophages and promote expression of ABCA1, which mediates cholesterol efflux to apoAI. Results from the study in this issue by Li et al. (15) suggest that PPARγ may also inhibit cholesterol accumulation in macrophages through direct regulation of ABCG1, which has been implicated in cholesterol efflux to HDL. Activation of each of the PPARs with selective agonists also inhibits the expression of inflammatory markers in the artery wall. These findings reinforce potential use of PPAR agonists as antiatherosclerotic therapies. PTLP, phospholipid transfer protein.
Differential effects of PPAR family members on the development of atherosclerosis in mice
In the current issue of the JCI, Li et al. (15) compare the effects of PPARγ, PPARα, and PPARβ ligands on the development of atherosclerosis in LDLR–/– mice. They observed profound atheroprotective effects of the PPARα ligand GW7647, comparable to that previously observed for the PPARγ agonists rosiglitazone and GW7845 (5). In contrast, no change in lesion development was observed in mice treated with PPARβ ligand. Beneficial metabolic effects of PPARα ligand included reduced weight gain, reduced insulin levels, and decreased levels of VLDL and LDL fractions. No significant changes were observed with PPARβ ligand. This shows that the ability to improve plasma lipid profiles and increase insulin sensitivity are likely to be major factors in the effects of PPARα and γ agonists on atherosclerosis observed in diabetic patients and hypercholesterolemic mice.
Li et al. (15) further explored the effects of PPARα and PPARβ agonists on gene expression in atherosclerotic mice. Each of the PPAR ligands was found to repress the expression of inflammatory markers in the artery wall even though PPARβ did not reduce lesion formation. PPARγ ligand increased the expression of CD36, LXRα, and ABCA1 and promoted cholesterol efflux in cultured macrophages. However, at the end of the study, ABCA1 expression was not induced by either ligand, and LXRα expression was induced only by the PPARα ligand. PPARγ did activate LXRα and ABCA1 at earlier stages. Thus, neither anti-inflammatory activity nor the ability to regulate LXR correlated well with the anti-atherosclerotic effects.
Effects of PPAR family members on macrophage foam-cell formation
To further explore the mechanism of PPAR actions in atherogenesis, Li et al. (15) developed a novel approach for evaluating macrophage foam-cell formation in vivo. This approach involved the adoptive transfer of peritoneal macrophages from various PPAR null mice into the peritoneal cavity of LDLR–/– mice fed a cholesterol-rich diet. This innovative assay provides a new model for macrophage lipid uptake although further validation is needed to clarify whether the assay faithfully reflects the actions of artery wall macrophages during atherogenesis. Nevertheless, Li et al. found that PPARα and PPARγ ligands inhibited lipid accumulation in a receptor-dependent manner, pointing to direct protective mechanisms within the macrophage. PPARγ ligand also markedly reduced cholesterol esterification in macrophages. This newly appreciated activity may be a significant contributor to the atheroprotective effects of PPARγ agonists.
The authors also investigated the potential role of LXRs in the antiatherogenic effects of PPAR ligands. LXR is a key transcriptional regulator of ABCA1 and ABCG1 expression and plays a major role in protection against cellular lipid overload, as deduced from studies using LXR-deficient macrophages and synthetic ligands (16–18). The results of Li et al. (15) suggest that the inhibition of foam-cell formation observed with PPARα ligands requires the expression of LXRs whereas inhibition by PPARγ ligands does not. Interestingly, the authors found that the ability of rosiglitazone to inhibit peritoneal foam-cell formation in WT and LXR null macrophages correlated with a modest increase in ABCG1 but not ABCA1 expression. Thus the ability of PPARγ to promote efflux independently of LXR may involve direct effects on efflux transporters (Figure 1). ABCG1 was recently shown to increase cholesterol efflux from macrophages to HDL, but not apoAI (19) and ex vivo macrophages from PPARγ ligand–treated mice showed increased cholesterol efflux to HDL, consistent with this mechanism. However, the definitive role of ABCG1 in lipid metabolism and atherosclerosis in vivo has not yet been established. Furthermore, the effects of PPAR agonists on ABCG1 expression in macrophages are small compared to those of LXR agonists (18), and the possibility that ABCG1 might be a direct target of PPARγ remains to be explored. In the future, it will be of interest to determine whether the atheroprotective effects of PPAR agonists are lost in mice lacking ABCG1.
Conclusions
The study by Li et al. (15) provides new insights into pathways regulating macrophage lipid accumulation and rounds out the family picture of PPARs in atherosclerosis (Figure 1). Both PPARα and PPARγ ligands were shown to protect against atherosclerosis in LDLR–/– mice and inhibit macrophage foam-cell formation. In contrast, the authors did not observe any effect from PPARβ activation. Given the discrepancies between PPARβ agonist effects in mice and primates, however, the possibility that PPARβ ligands may have beneficial effects on cardiovascular disease in humans is not excluded by the present study. Finally the work of Li et al. emphasizes that PPAR agonists are likely to exert their antiatherosclerotic properties by multiple mechanisms, including improving systemic lipid levels, improving insulin resistance, and inhibiting the accumulation of macrophage foam cells.
Footnotes
See the related article beginning on page 1564.Nonstandard abbreviations used: LDLR–/–, LDL receptor–deficient; LXR, liver X receptor.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- Lusis, AJ. Atherosclerosis. Nature. 2000. 407:233-241.
- Castrillo, A, Tontonoz, P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 2004. 20:455-480.
- Marx, N, Duez, H, Fruchart, JC, Staels, B. Peroxisome proliferator-activated receptors and atherogenesis: regulators of gene expression in vascular cells. Circ. Res. 2004. 94:1168-1178.
- Chawla, A, et al. A PPARgamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 2001. 7:161-171.
- Li, AC, et al. Peroxisome proliferator–activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor–deficient mice. J. Clin. Invest. 2000. 106:523-531.
- Collins, AR, et al. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001. 21:365-371. View this article via: PubMed
- Lee, CH, et al. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science. 2003. 302:453-457.
- Tordjman, K, et al. PPARα deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J. Clin. Invest. 2001. 107:1025-1034.
- Duez, H, et al. Reduction of atherosclerosis by the peroxisome proliferator-activated receptor alpha agonist fenofibrate in mice. J. Biol. Chem. 2002. 277:48051-48057.
- Rubins, HB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999. 341:410-418.
- Oliver (Jr), WR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. U. S. A. 2001. 98:5306-5311.
- Tontonoz, P, Nagy, L, Alvarez, JG, Thomazy, VA, Evans, RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998. 93:241-252.
- Chinetti, G, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001. 7:53-58.
- Ricote, M, Li, AC, Willson, TM, Kelly, CJ, Glass, CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998. 391:79-82.
- Li, AC, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J. Clin. Invest. 2004. 114:1564-1576. doi:10.1172/JCI200418730.
- Joseph, SB, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. U. S. A. 2002. 99:7604-7609.
- Tangirala, RK, et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 2002. 99:11896-11901.
- Venkateswaran, A, et al. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. J. Biol. Chem. 2000. 275:14700-14707.
- Wang, N, Lan, D, Chen, W, Matsuura, F, Tall, AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2004. 101:9774-9779.